
全文简介
析氧反应(OER)是水分解过程中限制性能的步骤。原位电化学可以诱导各种OER电催化剂的表面重构,动态形成反应位点,但代价是阳离子浸出速度快。因此,同时提高催化活性和稳定性仍然是一个重大挑战。在此,我们使用可扩展的阳离子缺陷驱动的溶解方法将均相掺杂的钴酸盐前驱体非原位重构为Ir/CoO/钙钛矿异质结(SCI-350),作为高活性且稳定的OER电极。SCI-350催化剂在1MKOH的10mAcm–2下表现出240mV的低过电位,在实际电解中表现出优异的耐久性超过150h。出色的活性初步归因于电荷积累的电化学表面积呈指数增加,从3.3mFcm–2增加到175.5mFcm–2。此外,密度泛函理论计算结合先进的光谱学和18O同位素标记实验证明了SCI-350上O-O偶联的三重氧交换动力学,增强了金属-氧杂化,并参与了晶格氧氧化。本工作为在不牺牲耐久性的情况下构建高活性氧化物OER电催化剂提供了一种有前景且可行的策略。
结果与讨论
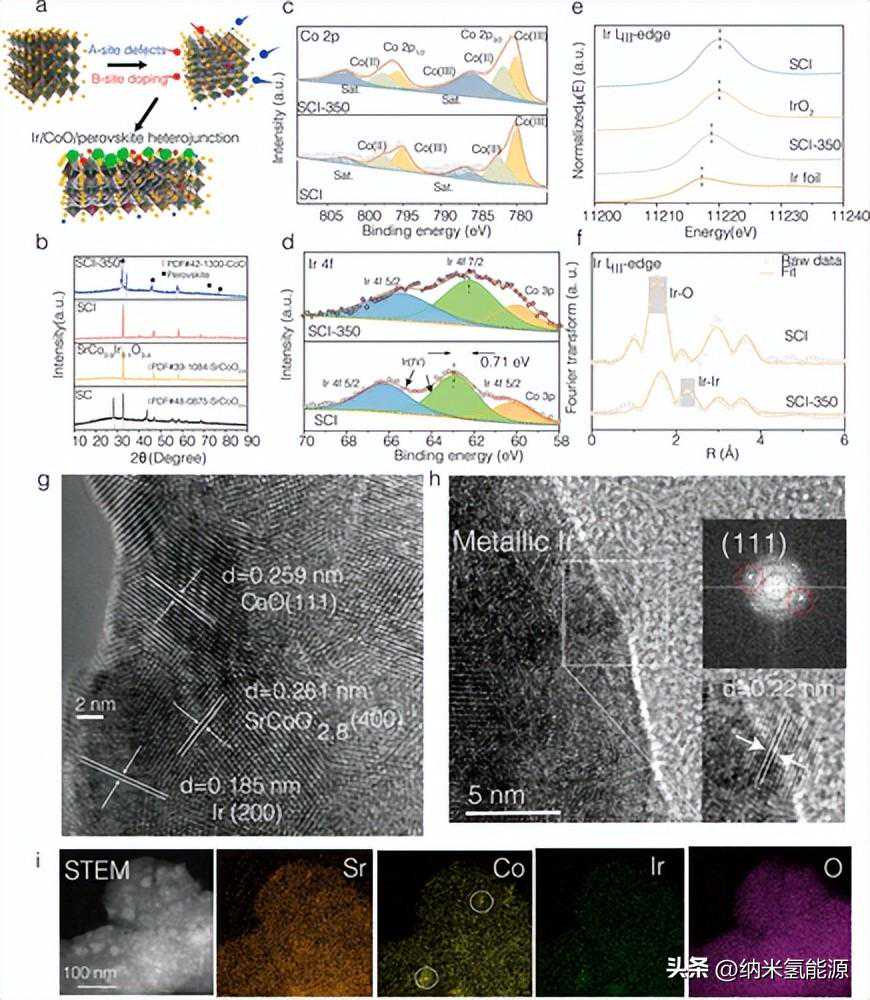
图1(a)SCI-350电催化剂的制作原理图。红色代表红色;绿色礼品公司;黄色代表Sr;蓝色为A-site;粉红色(小)代表0;绿色的锥面体表示CoO;红色三角面体呈现Ir团簇。(b)各种电催化剂的XRD谱图。(c,d)SCI和SCI-350的Co2p和Ir4fXPS光谱。(e)Ir箔、IrO2、SCI和SCI-350在Irliii边缘的XANES光谱。(f)SCI和SCI-350的k3加权IrLIII-edgeEXAFS光谱。(g,h)SCI-350的HR-TEM图像(金属Ir的PDF卡号为PDF#06-0598)。(i)SCI-350高角度环形暗场扫描透射电子显微镜(HAADF-STEM)和能量色散x射线(EDX)成像图像。
图1a描述了电催化剂在制造过程中的成分演变示意图。同时具有Ir掺杂和A位缺陷(SCI-350)的材料呈现出CoO和钙钛矿晶格畸变的共存(图1b)。此外,我们使用X射线光电子能谱(XPS)研究了电催化剂在制备过程中的化学状态演变(图1c,d和S4)。图1c,d分别显示了SCI和SCI-350的Ir4f和Co2p光谱。还原后,SCI-350的Ir结合能峰位置向下移动了∼0.71eV,表明Ir的平均氧化态降低。同时,SCI中Co的平均价态为∼2.61,而SCI-350中为∼2.50。这些结果表明,还原后Co的氧化态降低。通过X射线吸收光谱(XAS)进一步表征了Ir在各种电催化剂中的整体氧化态,如图1e所示。还给出了Ir氧化态为∼4+的SCI光谱和金属Ir参比。SCI-350的光谱位于中间,表明大部分Ir阳离子被大大还原。SCI和SCI-350的实验和拟合傅里叶变换(FT)IrL3边缘扩展X射线吸收精细结构(EXAFS)光谱如图1f所示(原始数据为点线,拟合曲线为红线)。HAADF-STEM(图1i)模式下的EDX映射结果显示了CoO纳米颗粒的偏析和固定在钙钛矿载体上的高度分散的Ir。
性能测试
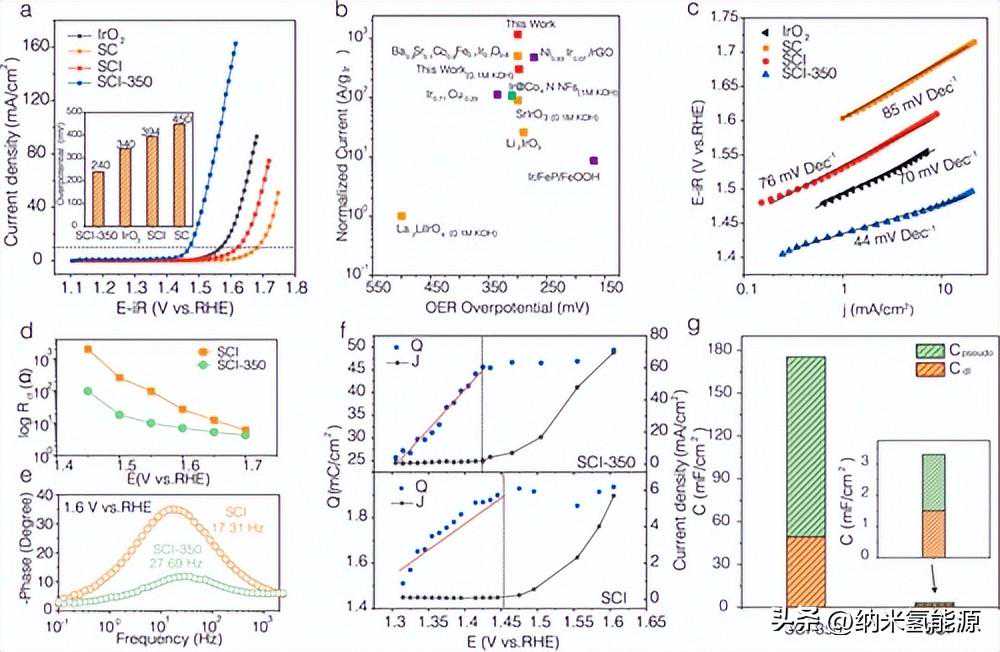
图2(a)不同电催化剂在1MKOH电解质中的LSV曲线。(b)代表性Ir基氧化物电催化剂的IrMA比较。电催化剂在1MKOH中测量,具体符号如图所示。(c)塔菲尔斜坡地块。(d)总电荷转移电阻(Rtotal)对外加电位的响应。(e)在1.6VvsRHE下的EIS-Bode图。(f)脉冲伏安法计算的QvsE和JvsE图。(g)伪电容与双层电容的比较。
使用三电极配置的线性扫描伏安法(LSV)评估各种电催化剂的OER性能。SCI-350在10mAcm–2(η10)的几何电流密度下具有240mV的低OER过电位,明显低于基准IrO2(340mV)、SCI(394mV)和SC(450mV),表明SCI-350具有卓越的OER活性(图2a)。SCI-350的OER活性在表观活性和归一化Ir质量活度(MA)方面均优于本研究中的其他催化剂和大多数最近报道的Ir基电催化剂(图2b)。为了准确量化电催化剂表面的总累积电荷(双电层电容和赝电容),进行了脉冲伏安法测量(图S17)。电催化剂最初设置为特定的正电位(1.305至1.605V),然后突然切换到开路电位(OCP)。对相应的阴极瞬态电流峰值进行监测和积分,以确定累积电荷。图2f总结了累积正电荷(Q)、OER电流密度(J)和外加电位(E)之间的关系,所有电催化剂都表现出相似的Q-E和J-E曲线形状。所有曲线都表现出两个不同的电位区域,由OER的起始电位隔开。在较低的电位下,OER未初始化,电流主要由表面电荷贡献,电催化剂在Q和E之间表现出线性关系。在达到OER起始电位后,OER电流开始增加,并且随着施加电位的增加,累积电荷几乎变得恒定。线性区域中(∂Q/∂E)的斜率可用于计算电催化剂的电荷存储容量,SCI-350的电容值为175.5mFcm–2,SCI的电容值为3.3mFcm–2(图2g)。因此,SCI-350和SCI的赝电容分别为126.3和1.8mFcm–2。大量的电荷积累是增加反应物表面覆盖率的原因,这进一步促进了OER动力学。
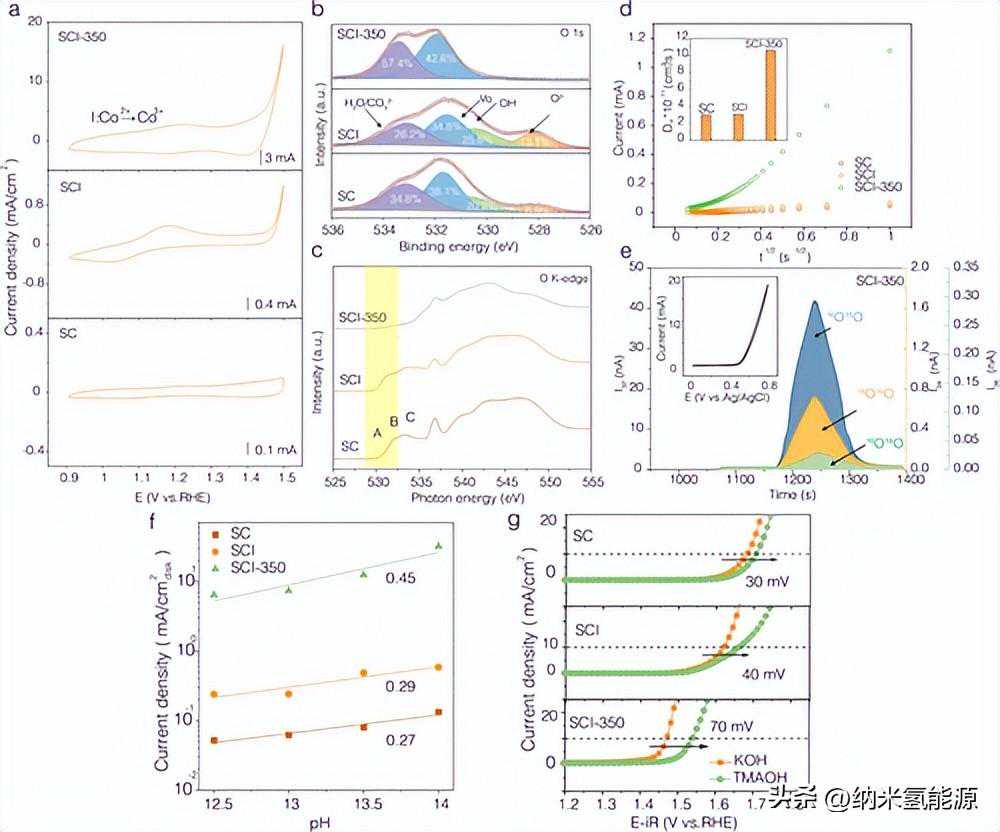
图3(a)在1MKOH中以5mV/s的扫描速率在Co氧化还原电位范围内的CV曲线。(b)O1sXPS光谱。(c)OK-edgeXANES图。(d)各种电催化剂的氧扩散系数实验。(e)16O2(I32)、16O18O(I34)和18O2(I36)的DEMS信号。插图显示了DEMS测量期间的循环伏安图。(f)1.5V时各种电催化剂的电流密度与RHE的关系随pH值的变化。(g)不同电催化剂在1MKOH和1MTMAOH电解液中的极化曲线。
由于电催化剂中的氧态与O-O偶联过程密切相关,因此使用XPS研究了O在表面的化学化合价。还原过程消除了晶格氧对应的峰,增加了氧空位的浓度。同时,H2O的面积比从26.2%增加到57.4%,表明材料的亲水性增强,有利于实现更快的OER动力学。在6MKOH电解质和氧空位激增量(图S20a)中进行CV实验,并使用计时电流法测定SC,SCI和SCI-350的氧离子扩散系数(DO)(图3d,S20b–d和表S7)。计算出SCI在室温下的DO值为3.08×10–11cm2s–1,与SC相当。值得注意的是,SCI-350具有更高的扩散系数,DO=10.58×10–11cm2s–1,是SCI的三倍以上(表S7)。SCI-350中的氧离子扩散加速被认为与氧空位浓度的增加有关。因此,通常期望在OER操作期间通过LOM途径促进晶格氧参与。此外,大量空氧位点的存在可以提高电导率,这有利于OER的电荷转移。为了验证我们的假设,我们对18O同位素标记进行了预处理,然后在18O标记的SCI-350上进行了原位差分电化学质谱(DEMS)测量。结果显示m/z=32、m/z=34和m/z=36的显着信号(图3e),表明在析氧过程中存在16O2、16O18O和18O2。这一结果表明,SCI-350遵循LOM机制,该机制共同激活了直接晶格氧偶联或晶格氧和OH基团氧的共同参与。通常,LOM之后的电催化剂表现出pH依赖性活性。提高pH值可以改变吸附中间体的能量,也可以增加表面覆盖率或OH浓度,从而增加OER活性。我们的实验研究证实,当pH值从12.5增加到14时,所有样品的OER活性都有所增加(图S21),这表明OER动力学和LOM参与与pH值有关。图3f进一步比较了所有电催化剂在1.50V与RHE下OER活性随pH值的变化,斜率为[ρ=(∂logi/∂pH)E]SCI-350、SCI和SC分别为0.45、0.29和0.27。因此,形成的Ir/CoO/钙钛矿异质结具有较高的晶格氧参与OER的倾向。另一方面,通过LOM的OER涉及负氧物质的形成[过氧化样(O2−2)和可被四甲基铵阳离子(TMA+)捕获的超氧样(O2–)]。因此,我们比较了不同电催化剂在1MKOH和TMAOH溶液中的OER活性。如图3g和S22所示,SCI-350在10mAcm–2时显著增加OER过电位70mV,其Tafel斜率也增加了24mVdec-1,这主要是由于LOM的抑制。
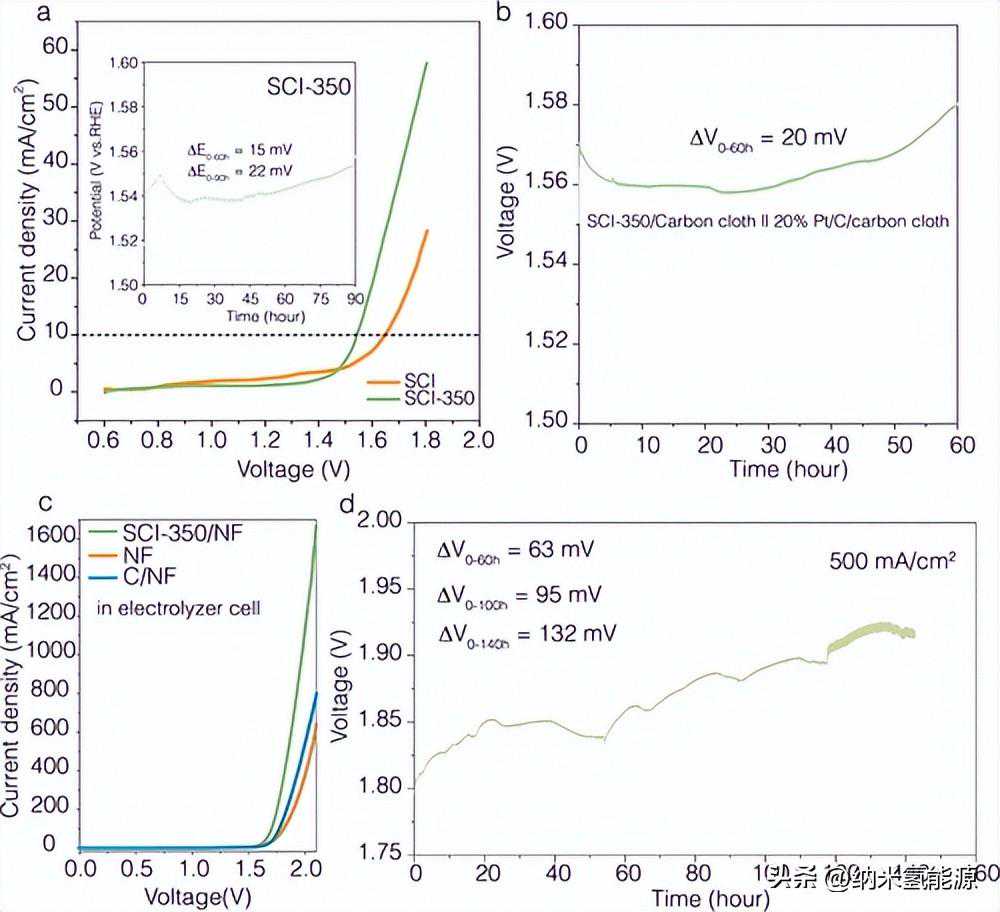
图4(a)装有OER电催化剂的电解槽在1MKOH溶液中的LSV曲线。插图显示了在1MKOH溶液下三电极配置下的耐久性测试。(b)1MKOH溶液下双电极构型的稳定性测试。(c)6MKOH溶液下SCI-350/NF、裸NF和C/NF在水电解槽中的LSV曲线(电压为iR校正)。(d)电解水单电池耐久性试验。电池电压(电压经过iR校正)为500mAcm–2。电压变化值(ΔV0–60h)是60小时内最高电压和最低电压之间的差值。
最后,SCI-350在恒电流模式下以10mAcm–2进行耐久性测试90h,OER电位衰减为22mV(图4a插图)。然后使用双电极设置来研究SCI和SCI-350催化剂的稳定性(图4a)。为了了解SCI-350的阳离子浸出效应,我们使用ICP来量化电解过程中电解液中溶解金属阳离子的浓度。我们对SCI-350在恒电流模式下以20mAcm–2进行了10小时的独立耐久性测试(图S30a),并在不同的电解时间进行了ICP测量。如图S30b,c所示,Sr、Co和Ir的浸出速率分别为105.14、0.94和6.21ppbh–1,表明催化活性元素Ir和Co的损失比Sr慢得多。
结论
本研究报道了一种用于制备Ir/CoO/钙钛矿异质结作为活性和持久OER催化剂的非原位重建方法。同时考虑了A位缺陷和溶蚀设计原则,以实现催化剂的最佳组成。合成的SCI-350在10mAcm–2时表现出240mV的低过电位,并显示出用于实际水分解电解的良好潜力,在150小时内保持500mAcm–2的稳定电流密度。详细的电化学分析证实,电荷积累能力增加了一个数量级(从3.3mFcm–2增加到175.5mFcm–2)。此外,XAS、XPS和原位拉曼数据的结合表明存在大量空氧,这促进了OER过程中晶格氧扩散和加速氧交换。18O同位素标记的DEMS和DFT计算进一步证明了O-O耦合的晶格氧活性,这是由于金属-氧共价的增强和O2p中心相对于费米能级的上移。我们的研究结果将为通过LOM途径合理设计高效氧化物基OER催化剂提供启示。
参考文献
HongquanGuo,YanlingYang,GuangmingYang,XiaojuanCao,NingYan,ZhishanLi,EmilyChen,LinaTang,MeilanPeng,LeiShi,ShunjiXie,HuabingTao,ChaoXu,YinlongZhu,XianzhuFu,YuanmingPan,NingChen,JinruLin,XinTu,ZongpingShao,(7),5007-5019.





